molchemist is a Typst package for rendering chemical structures from Molfile / SDF data and from SMILES strings.
It uses a Rust/WASM core to parse molecular graphs and generate alchemist ASTs, together with a companion WASM layout plugin for SMILES 2D coordinate generation. Molfile / SDF parsing is powered by sdfrust, SMILES parsing is based on opensmiles, SMILES 2D coordinate generation uses CoordgenLibs, and the final rendering is handled by the declarative drawing engine of alchemist.
Third-party license notices and bundled example-data provenance are collected in THIRD_PARTY_NOTICES.md.
Usage
Import render-mol for Molfile/SDF inputs, or render-smiles for SMILES inputs.
#import "@preview/molchemist:0.1.2": render-mol, render-smiles
// Read your molecule data
// Example: https://pubchem.ncbi.nlm.nih.gov/compound/93406
#let mol-data = read("Structure2D_COMPOUND_CID_93406.sdf")On Typst 0.15.0 and later, you may also pass path("Structure2D_COMPOUND_CID_93406.sdf") directly to render-mol; molchemist will read the file inside the package. The examples in this README use read(...) for compatibility with older Typst versions.
The bundled documentation and README screenshots use small PubChem-derived example structures. See THIRD_PARTY_NOTICES.md for source URLs and NCBI data-usage notes.
For SMILES, molchemist generates a 2D layout internally before sending the structure to alchemist.
// Example: https://pubchem.ncbi.nlm.nih.gov/compound/896
#render-smiles("CC(=O)NCCC1=CNC2=C1C=C(C=C2)OC", abbreviate: true)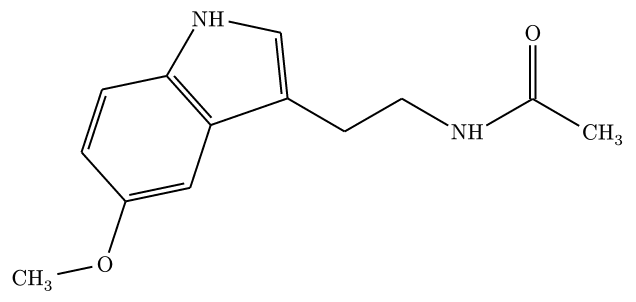
Adding Annotations
You can overlay arrows and labels on top of a rendered molecule with the annotations argument. molchemist provides helpers for atom-level, bond-level, and molecule-level annotations without leaving the package API.
#import "@preview/molchemist:0.1.2": (
render-smiles,
atom-anchor,
bond-anchor,
molecule-anchor,
callout-annotation,
arrow-annotation,
)
#render-smiles(
"OCCc1c(C)[n+](=cs1)Cc2cnc(C)nc(N)2",
abbreviate: true,
annotations: (
callout-annotation(
atom-anchor(6, anchor: "north"),
[cationic center],
side: "north-east",
),
callout-annotation(
bond-anchor(3, anchor: "50%"),
[aromatic bond],
side: "north-west",
),
arrow-annotation(
molecule-anchor(anchor: "east"),
(rel: (2.6, 0), to: molecule-anchor(anchor: "east")),
label: [reaction direction],
label-offset: (0, -0.45),
label-anchor: "north",
),
),
)Use callout-annotation(...) for most explanatory labels, arrow-annotation(...) for free arrows, and label-annotation(...) for low-level labels. atom-anchor(...) targets a specific atom, bond-anchor(...) targets a specific bond, and molecule-anchor(anchor: "center") attaches to the molecule as a whole. Callouts are intentionally restrained for publication figures: unboxed labels, thin monochrome leader lines, no arrowheads by default, and enough clearance from both the label text and the chemical structure. Placement presets such as side: "north-east" cover the common cases, while label-at, leader-start, leader-end, leader-points, label-gap, and target-gap are available for small manual corrections when a paper figure needs precise spacing. For final figure-level adjustments, cetz-annotation((mol) => { ... }) exposes the generated molecule name for direct Cetz drawing. To discover the atom and bond indices for a molecule, enable the debug overlay with show-indices: true, show-indices: "atoms", or show-indices: "bonds". In abbreviated or skeletal mode, the overlay only labels elements that are actually rendered.
Publication Figure Guidance
For paper figures, prefer skeletal: true for hydrocarbon-heavy structures and abbreviate: true when heteroatom hydrogens or terminal groups should remain explicit. Use Full Mode mainly for small molecules or debugging, since explicit hydrogens can make dense structures hard to read.
Keep annotations minimal: use monochrome callout-annotation(...) labels, avoid arrowheads unless the line represents a process, and move labels outside the molecular graph. If a leader line visually resembles a chemical bond, increase target-gap, move the label with label-at, or route the line through leader-points.
Rendering Modes
molchemist supports three distinct rendering styles to suit your document’s needs:
1. Full Mode (Default)
Draws every single atom and bond explicitly exactly as defined in the source file, including all carbons and hydrogens.
Note: For complex molecules, text overlapping may occur. See Known Limitations for workarounds.
#render-mol(mol-data)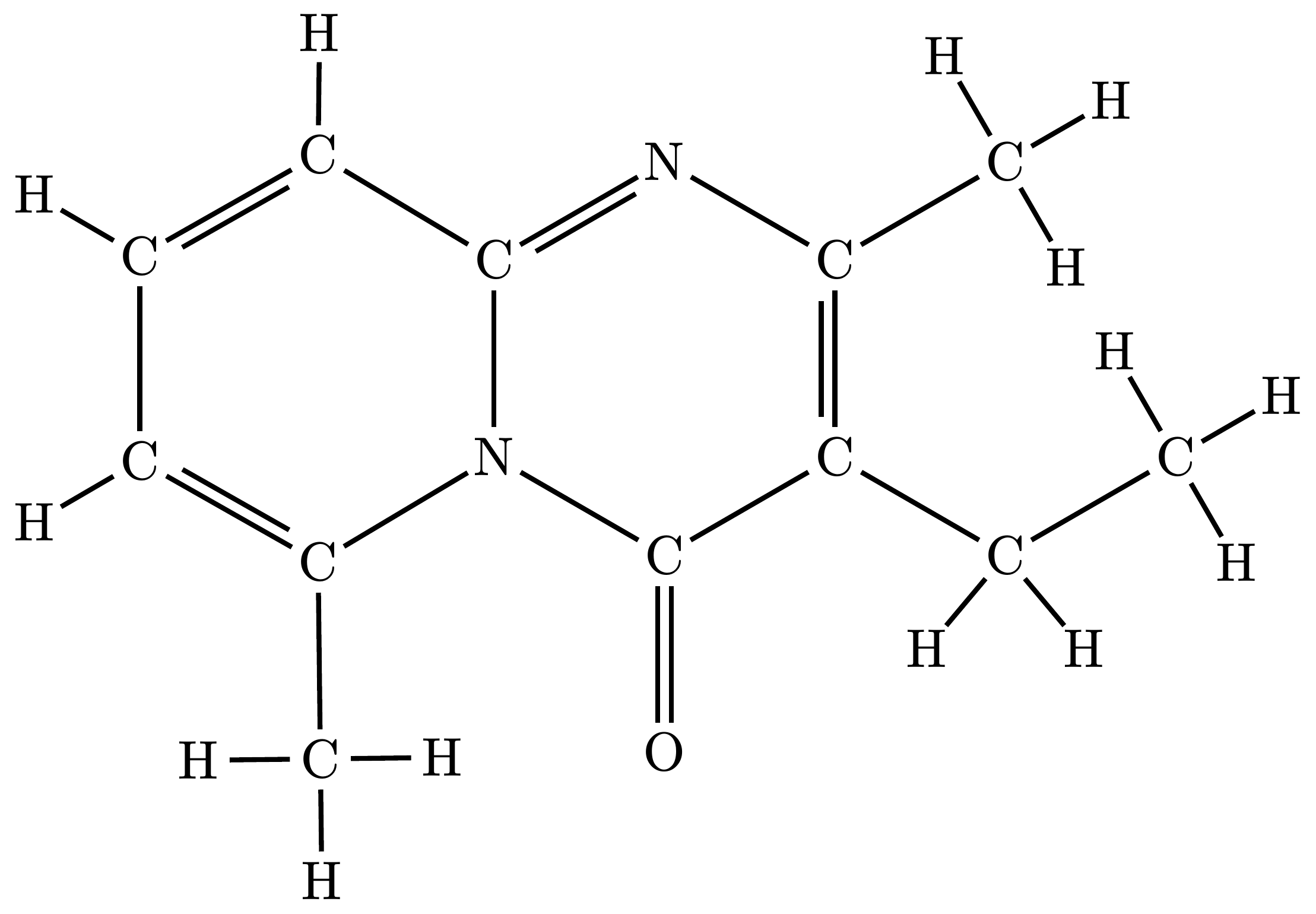
2. Abbreviated Mode
A standard chemical representation. It hides the carbon backbone, wraps explicit hydrogens into their parent heteroatoms (e.g., O + H becomes OH), and neatly formats terminal carbon groups (e.g., CH3).
#render-mol(mol-data, abbreviate: true)
3. Skeletal Mode
A pure skeletal formula. All backbone carbons and their attached hydrogens are completely hidden, leaving only the zigzag lines and heteroatoms.
#render-mol(mol-data, skeletal: true)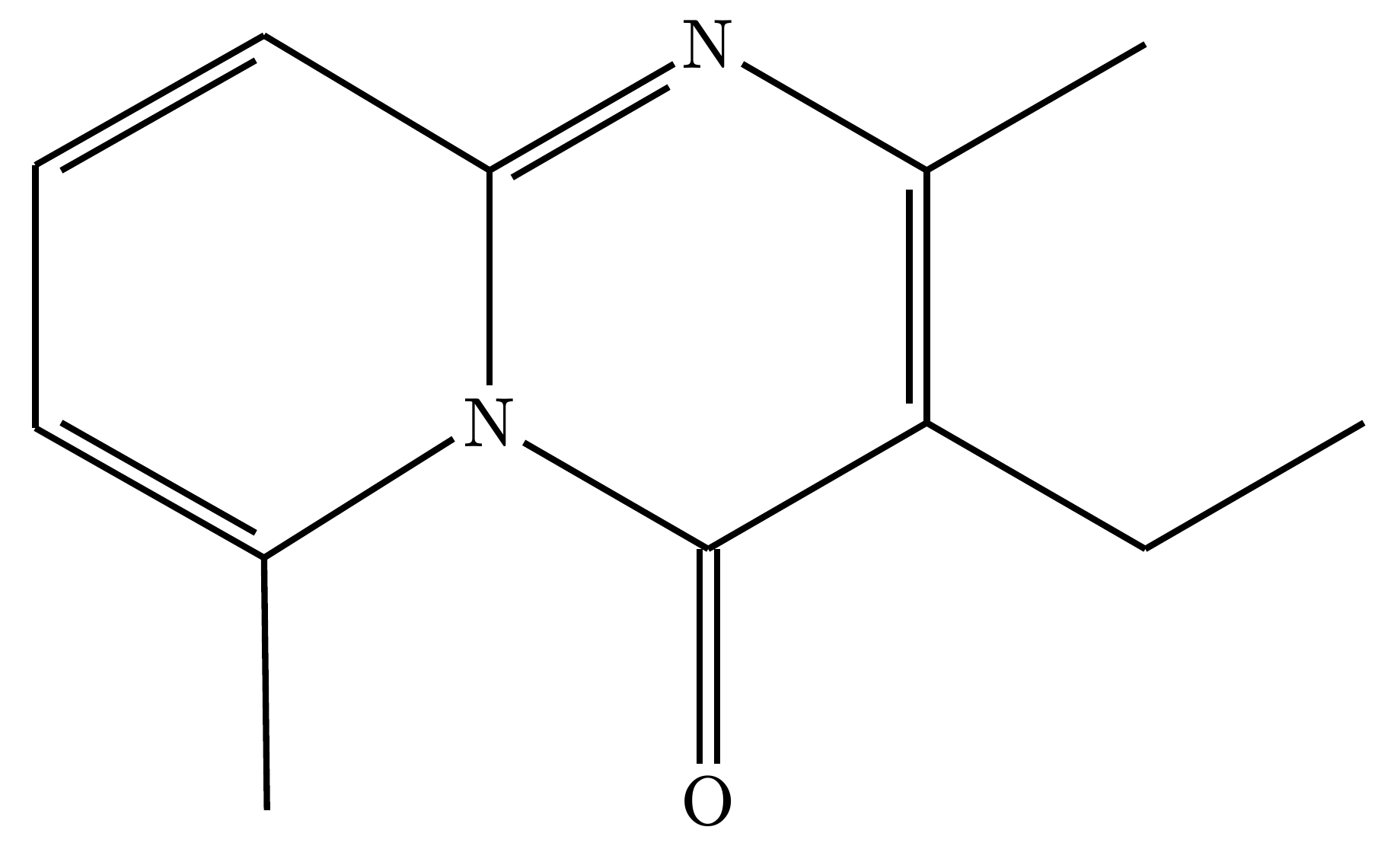
Customizing Appearance
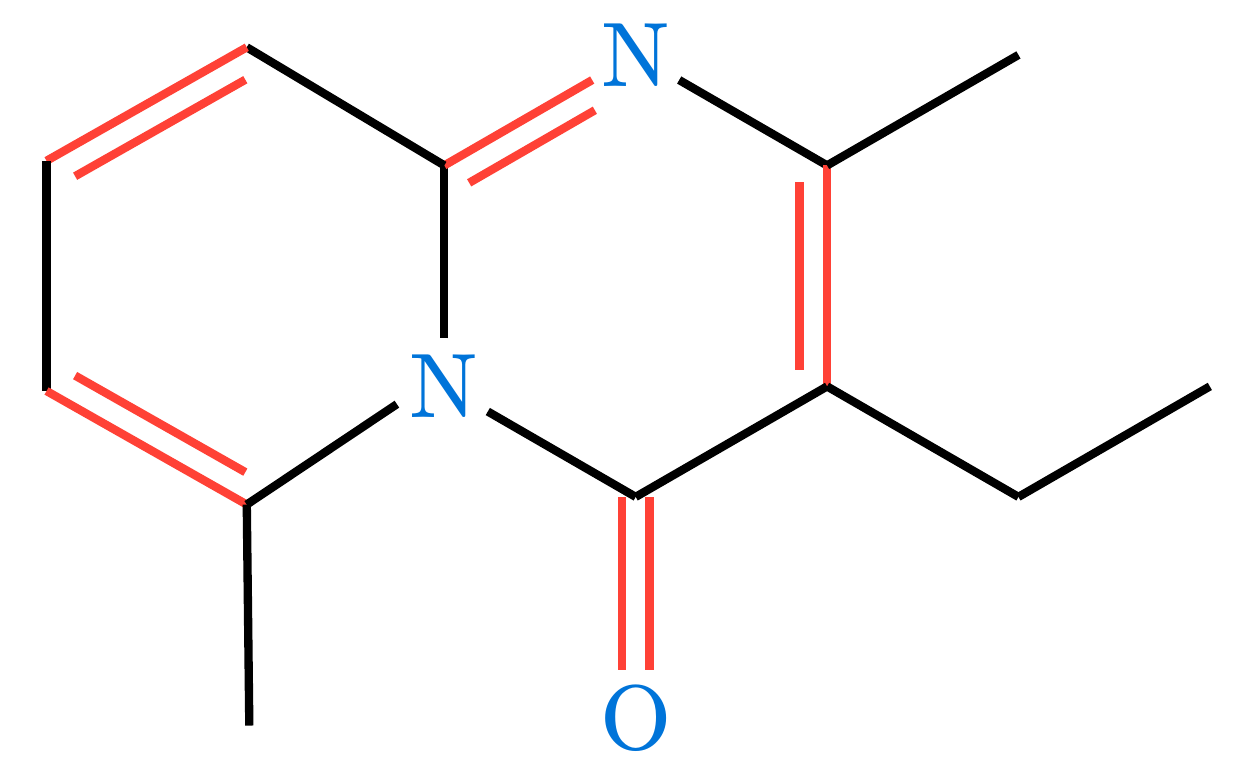
Under the hood, molchemist parses the graph and generates native alchemist elements. You can customize the look of your molecules by passing styling arguments via the config dictionary, which are passed directly to alchemist’s skeletize function.
#render-mol(
mol-data,
skeletal: true,
config: (
atom-sep: 2em,
fragment-margin: 2pt,
fragment-color: blue,
fragment-font: "New Computer Modern",
single: (stroke: 1pt + black),
double: (gap: 0.3em, stroke: 1pt + red)
)
)
Important Note on Configuration:
- Routing overrides: Because
molchemistmaps the exact 2D absolute coordinates from the source.sdf/.molfile,alchemist’s automatic routing configs (likeangle-increment,base-angle) are bypassed and have no effect. - Lewis Structures:
molchemistdoes not automatically infer or generate Lewis structures from SDF files, solewis-*configs are not applicable out of the box.
Advanced: Ejecting to Alchemist Code (Dump Mode)
If you need to manually fine-tune a molecule, add a specific Lewis structure, or integrate the structure into a larger custom alchemist drawing, you can use the dump parameter.
When dump: true is passed, molchemist will not render the molecule. Instead, it will output the generated native alchemist code block into your document. You can then copy, paste, and modify this code directly.
#render-mol(mol-data, dump: true)
Known Limitations
When rendering highly complex or dense molecules (e.g., polycyclic compounds, dense substituents) in the default Full Mode, you may encounter overlapping atoms or intersecting bonds. This occurs because the 2D absolute coordinates provided in the source .sdf/.mol files might not allocate enough physical space on the canvas to draw every explicit text label without collisions.
Recommended Workarounds:
- Use Abbreviated or Skeletal Mode: For complex organic structures, it is highly recommended to set
abbreviate: trueorskeletal: true. This hides redundant atoms, dramatically improving readability and preventing overlaps, which aligns with standard chemical drawing practices. Increase Bond Length: If you strictly require Full Mode, you can increase the distance between atoms to create more physical space for the text labels by adjusting the
atom-sepproperty in theconfigargument:// The default atom-sep is 3em #render-mol(mol-data, config: (atom-sep: 4.5em))
For SMILES input, the default render-smiles(...) mode expands implicit hydrogens into explicit H atoms so that full mode stays closer to the behavior of render-mol(...). Highly complex or dense molecules can still become visually busy in full mode, so abbreviate: true or skeletal: true will often produce a clearer result. The current implementation also supports tetrahedral @ / @@ centers and / / \ double-bond geometry as stereochemical depictions. Extended OpenSMILES chirality classes such as @AL, @SP, @TB, and @OH are accepted as well; because the current alchemist-based renderer does not have native glyphs for those geometries, they are preserved as stereo annotations below the rendered structure instead of wedge/dash depictions.
Feature Plan
- Native depiction for extended chirality: Extended OpenSMILES chirality classes such as allene (
@AL), square-planar (@SP), trigonal-bipyramidal (@TB), and octahedral (@OH) are currently preserved as textual stereo annotations. A future version may render these classes natively once the expected 2D depiction conventions and the requiredalchemistprimitives are clarified.
API Reference
Renderers
#render-mol(data, ..options)
#render-smiles(smiles, ..options)| Function | Input | Description |
|---|---|---|
render-mol | data: str, bytes, or Typst 0.15+ path | Renders Molfile or SDF data. Coordinates are read from the input. |
render-smiles | smiles: str | Parses SMILES, generates a 2D layout, and renders the result. |
Both renderers accept the same options:
| Option | Type | Default | Description |
|---|---|---|---|
abbreviate | bool | false | Folds common hydrogens and terminal groups into labels. |
skeletal | bool | false | Draws a skeletal formula. Overrides abbreviate. |
dump | bool | false | Returns generated alchemist source instead of rendering. |
config | dictionary | (:) | Passes visual settings directly to alchemist. |
annotations | annotation, array, none | none | Adds labels, arrows, or custom CeTZ overlays. |
show-indices | bool, str | false | Shows debug labels for annotation authoring. Use true, "all", "atoms", or "bonds". |
Anchors
Use anchors to target atoms, bonds, or the whole molecule from annotations.
| Function | Returns | Use |
|---|---|---|
atom-anchor(index, anchor: "mid") | anchor selector | Target a rendered atom. |
bond-anchor(index, anchor: "50%") | anchor selector | Target a rendered bond. |
molecule-anchor(anchor: "center") | anchor selector | Target the rendered molecule group. |
atom-ref(index) | str | Inspect the generated atom anchor name. |
bond-ref(index) | str | Inspect the generated bond anchor name. |
Annotations
Pass one annotation or an array of annotations to annotations.
| Function | Purpose |
|---|---|
callout-annotation(at, label, ..options) | External publication-style label with a thin leader line. |
arrow-annotation(from, to, ..options) | Free arrow overlay for process arrows or directional marks. |
label-annotation(at, label, ..options) | Free text label without a leader line. |
cetz-annotation(body, ..options) | Low-level CeTZ overlay. The callback receives the generated molecule name. |
Common callout-annotation controls include side, label-at, leader, leader-start, leader-end, leader-points, label-gap, and target-gap. Use these when a final figure needs precise spacing.
Use cetz-annotation as the escape hatch for advanced figure polishing:
#render-smiles(
"c1ccccc1",
skeletal: true,
annotations: cetz-annotation(mol => {
import cetz.draw: *
content((to: (name: mol, anchor: "north"), rel: (0, 0.45)))[benzene]
}),
)License
The molchemist package code is distributed under the MIT License. See LICENSE for details.
Redistributed third-party code and bundled example-data provenance, including PubChem-derived SDF/example images, are documented separately in THIRD_PARTY_NOTICES.md.